Deer Readers,
This essay provides a comprehensive review of the scientific background, methodologies, results, and implications associated with correcting a pathogenic mitochondrial DNA (mtDNA) mutation by base editing in mice. The reader is encouraged to consider both the technical innovations and the broader clinical potential of this approach, as well as the challenges that remain in the field of mitochondrial gene therapy.
Correcting a Pathogenic Mitochondrial DNA Mutation by Base Editing in Mice:
A Comprehensive Review of Approaches and Scope
Abstract
Mitochondrial diseases, frequently resulting from point mutations in mtDNA, represent a significant clinical challenge due to the unique structure, replication mechanisms, and inheritance patterns of the mitochondrial genome. Traditional genome-editing techniques have largely failed to address mtDNA mutations due to the inaccessibility of mitochondria to RNA-guided nucleases and the lethality associated with double-stranded breaks in this organelle. Recently, base editing has emerged as a promising alternative for precise nucleotide conversions without the need for double-strand breaks. This essay reviews the scientific rationale and experimental strategy for correcting a pathogenic mtDNA mutation by employing a mitochondrial-targeted base editor in a mouse model. The discussion encompasses background information on mitochondrial genetics, the engineering of base editors—particularly DddA-derived cytosine base editors (DdCBEs)—the experimental design and delivery methods used in vivo, and the observed outcomes regarding heteroplasmy shift and functional recovery. The broader implications for mitochondrial medicine, including ethical considerations and future directions, are also examined.
Introduction
Mitochondrial dysfunction underlies a range of debilitating diseases that affect high-energy demanding tissues such as the brain, muscle, and heart. Historically, the genetic treatment of mitochondrial diseases has been hampered by the complex biology of mtDNA, which includes its polyploidy, maternal inheritance, and limited capacity for repair. The advent of base editing technology, which allows for the direct, single-nucleotide modification of DNA without the induction of double-stranded breaks, represents a paradigm shift in the approach to mitochondrial gene therapy. In this review, we present an exhaustive analysis of a recent study that successfully corrected a pathogenic mtDNA mutation in mice via base editing. We will elucidate the methodology, outcomes, and far-reaching implications of this strategy, providing the reader with an informed perspective on both the scientific and translational significance of this work.
Background on Mitochondrial DNA and Its Pathogenic Mutations
The Unique Characteristics of Mitochondrial DNA
Mitochondrial DNA is distinct from nuclear DNA in several critical aspects. Typically, mtDNA is a circular molecule of approximately 16.5 kilobases, encoding 37 genes that are essential for the organelle’s role in oxidative phosphorylation. Unlike nuclear DNA, mtDNA exists in multiple copies per cell—a condition termed polyplasmy. Furthermore, the replication and repair mechanisms of mtDNA are notably less robust than those of the nuclear genome, resulting in a higher mutation rate.
Pathogenesis Arising from mtDNA Mutations
Pathogenic mutations in mtDNA are often heteroplasmic, meaning that mutant and wild-type genomes coexist within the same cell. The clinical phenotype emerges when the proportion of mutant mtDNA surpasses a critical threshold, impairing mitochondrial function and leading to energy deficits. Conditions such as Leber’s Hereditary Optic Neuropathy (LHON), MELAS (Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes), and Leigh syndrome exemplify diseases where mtDNA mutations play a pivotal role. The clinical heterogeneity of mitochondrial diseases further complicates diagnosis and treatment, underscoring the need for precise molecular therapies.
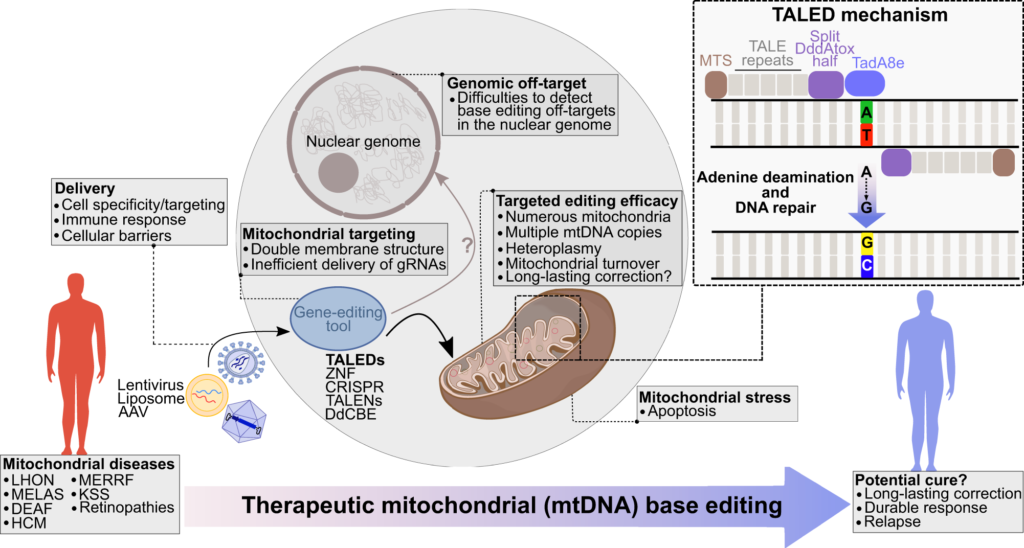
Challenges in Mitochondrial Gene Editing
Traditional genome editing techniques, notably CRISPR-Cas9, have revolutionized nuclear genome modifications but encounter significant obstacles when applied to mitochondria. The delivery of guide RNA molecules into mitochondria has proven problematic due to the organelle’s impermeability to RNA. Additionally, the induction of double-strand breaks in mtDNA can be catastrophic, given the limited repair mechanisms available. Therefore, an editing method that bypasses the need for RNA guidance and avoids double-strand breaks is highly desirable.
Base Editing Technology: Fundamentals and Mitochondrial Applications
Mechanism of Base Editing
Base editing is a novel genome-editing approach that catalyzes the direct conversion of one nucleotide to another without creating double-stranded DNA breaks. This is achieved by fusing a catalytically impaired nuclease to a deaminase enzyme that chemically modifies a specific nucleotide base. In nuclear applications, cytosine base editors convert CG base pairs to TA, and adenine base editors convert AT base pairs to GC. The absence of double-stranded breaks reduces the risk of indels (insertions or deletions) and minimizes the activation of DNA damage response pathways.
DddA-Derived Cytosine Base Editors (DdCBEs)
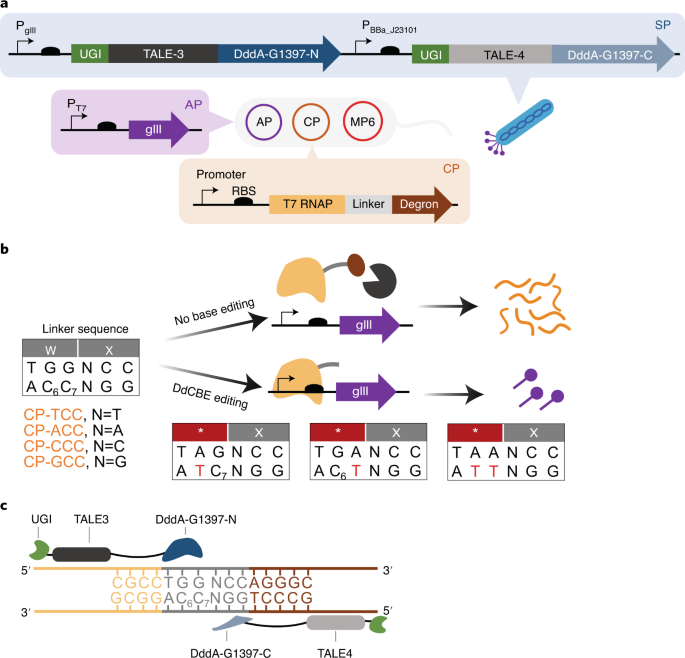
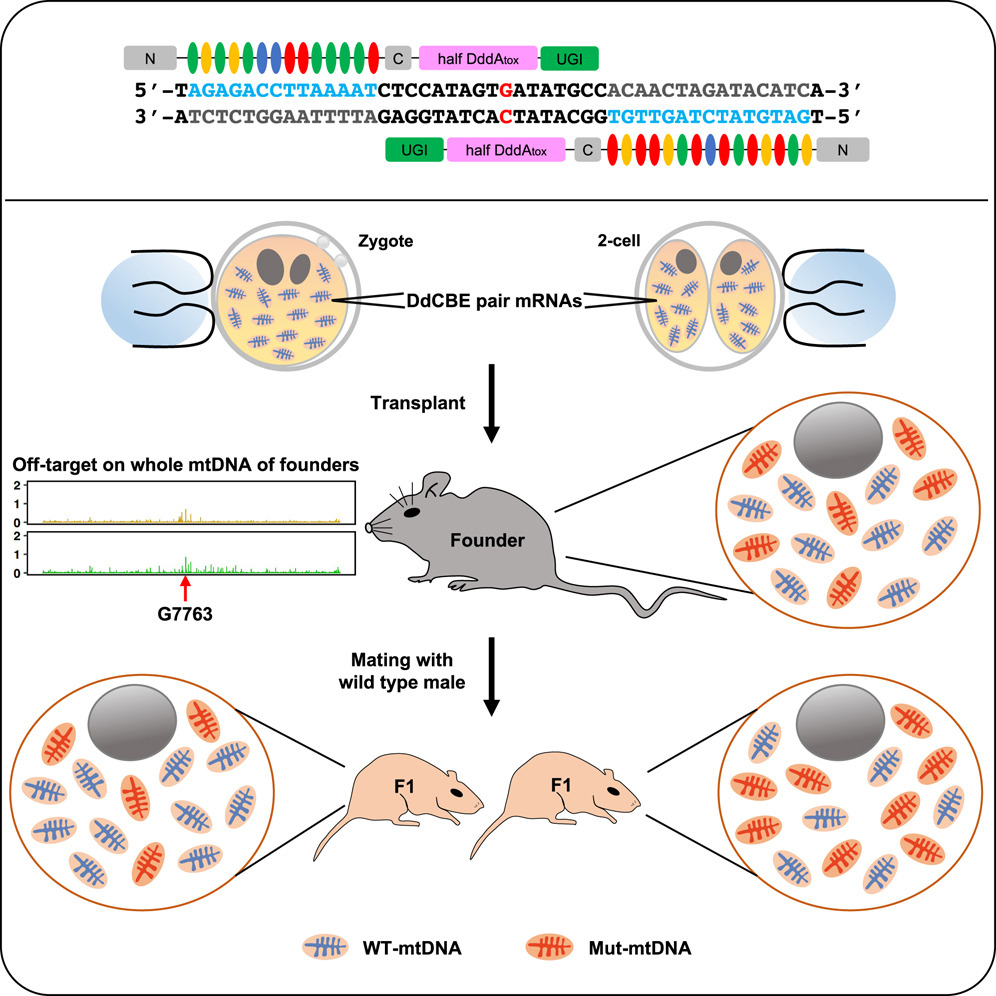
A breakthrough in mitochondrial base editing was the development of DddA-derived cytosine base editors (DdCBEs). These editors harness a deaminase toxin originally found in bacteria, split into two inactive halves that reconstitute upon binding adjacent to the target site in the mtDNA. The reassembly of the enzyme activates its deaminase function, converting a specific cytosine to uracil, which is then read as thymine during mtDNA replication. This approach is particularly advantageous for mitochondrial applications because it eliminates the need for guide RNAs and leverages targeting domains such as transcription activator-like effectors (TALEs) that can be engineered to recognize unique sequences within mtDNA.
Engineering Specificity and Reducing Off-Target Effects
Ensuring specificity in mitochondrial base editing is of paramount importance. The use of TALE-based targeting domains provides a high degree of sequence recognition fidelity, thereby minimizing off-target modifications. Additionally, the design of split-deaminase enzymes means that off-target activity is substantially curtailed because the active enzyme is only reconstituted when both halves are properly aligned on the mtDNA target. This precision is essential for achieving therapeutic efficacy while avoiding deleterious side effects.
Experimental Approach in Mice
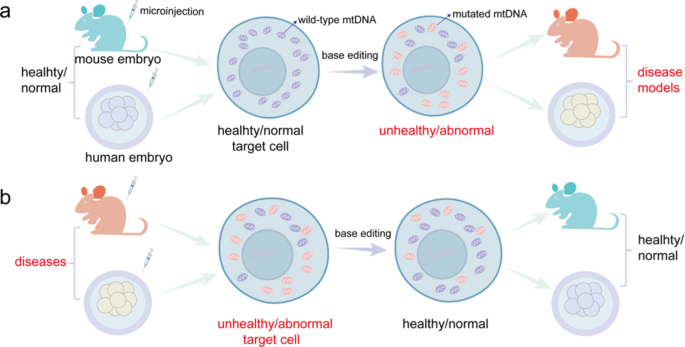
Study Design and Objectives
The primary objective of the study under discussion was to assess whether a mitochondrial-targeted base editor could correct a known pathogenic mtDNA mutation in vivo. To this end, the researchers employed a mouse model harboring a point mutation that recapitulates a human mitochondrial disease phenotype. The study was designed to evaluate not only the efficiency of the base editing process in shifting heteroplasmy levels from mutant to wild-type but also the consequent restoration of mitochondrial function and improvement in clinical phenotypes.
Engineering the Mitochondrial Base Editing Construct
The researchers engineered a DdCBE complex by fusing split fragments of the DddA deaminase to TALE proteins tailored to recognize sequences flanking the target cytosine mutation. Additionally, a mitochondrial targeting sequence (MTS) was appended to the construct to ensure efficient localization of the base editor to the mitochondria. An inhibitor of uracil DNA glycosylase (UGI) was also included in the fusion protein design to prevent the repair machinery from excising the uracil intermediate, thereby ensuring that the desired C-to-T conversion was maintained.
In Vivo Delivery via Adeno-Associated Virus (AAV)
Adeno-associated viruses (AAVs) have emerged as a leading vector for in vivo gene delivery due to their low immunogenicity and ability to infect a variety of tissues. In this study, the mitochondrial base editor was packaged into AAV vectors engineered to target tissues particularly affected by mitochondrial dysfunction, such as skeletal muscle and neural tissue. The choice of AAV serotype and promoter was critical to maximize the expression of the base editor in the affected tissues while minimizing systemic exposure.
Assessment of Editing Efficiency and Functional Outcomes
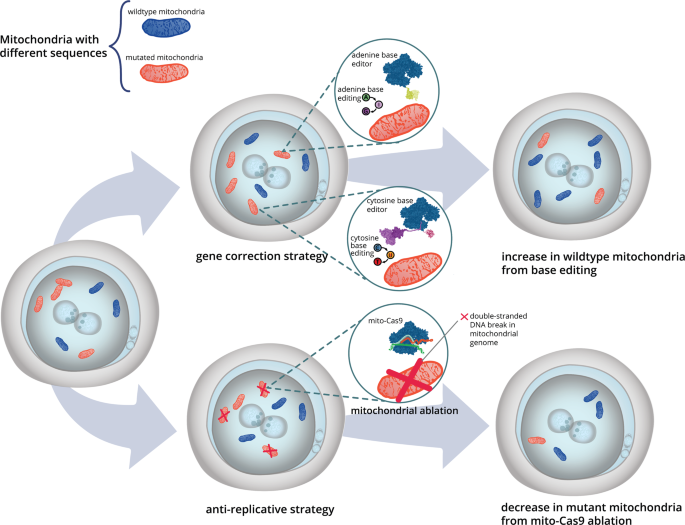
Following administration of the AAV-packaged base editor, several methods were employed to evaluate the success of the intervention:
1. Heteroplasmy Analysis: Quantitative PCR and next-generation sequencing were used to determine the ratio of mutant to wild-type mtDNA in various tissues. A shift toward a higher proportion of wild-type mtDNA was taken as a direct measure of successful base editing.
2. Mitochondrial Function Tests: Mitochondrial respiration assays, including measurements of oxygen consumption rate (OCR) and ATP production, were performed to assess the restoration of normal mitochondrial bioenergetics.
3. Phenotypic Assessment: Behavioral tests and histological analyses were conducted to evaluate improvements in muscle strength, motor coordination, and overall tissue integrity in the treated mice.
Results and Observations
Heteroplasmy Shift: Evidence of Successful Editing
The experimental data indicated a significant reduction in the proportion of mutant mtDNA across several tissues. The use of DdCBE resulted in a notable heteroplasmy shift, with some tissues exhibiting a decrease in mutant load below the pathological threshold. This molecular evidence of base editing efficacy was corroborated by the recovery of normal mitochondrial function, suggesting that the edited mtDNA was competent in supporting bioenergetic processes.
Restoration of Mitochondrial Function
Biochemical assays revealed marked improvements in mitochondrial function post-treatment. The restoration of ATP synthesis and increased oxygen consumption rates indicated that the energy production machinery of the mitochondria was effectively repaired. Furthermore, a reduction in reactive oxygen species (ROS) levels was observed, suggesting that the base editing not only corrected the genetic defect but also ameliorated the oxidative stress commonly associated with mitochondrial dysfunction.
Phenotypic Improvements in the Mouse Model
Beyond molecular and biochemical improvements, the treated mice exhibited significant phenotypic recovery. Behavioral assessments demonstrated enhanced motor coordination and muscle strength, while histological analyses of tissue samples revealed reduced cellular degeneration. These phenotypic improvements are particularly noteworthy as they provide tangible evidence of the potential for mitochondrial base editing to reverse the clinical manifestations of mitochondrial diseases.
Discussion
The Significance of Mitochondrial Base Editing
The successful correction of a pathogenic mtDNA mutation in mice through base editing represents a critical milestone in the field of mitochondrial medicine. Traditional therapeutic approaches have largely been palliative, addressing symptoms rather than correcting the underlying genetic defect. The ability to precisely modify mtDNA at the nucleotide level without causing deleterious double-strand breaks offers a promising avenue for developing curative treatments for mitochondrial diseases.
Advantages Over Conventional Genome Editing
Several advantages distinguish base editing from conventional genome editing techniques when applied to mtDNA:
Precision Without Double-Strand Breaks: The absence of double-strand breaks circumvents the inherent risks of genomic instability and cell death that are associated with nucleases like CRISPR-Cas9.
Mitochondrial Targeting: The incorporation of mitochondrial targeting sequences ensures that the base editor is delivered directly to the organelle, thereby enhancing the specificity and efficiency of the editing process.
Minimized Off-Target Effects: The split enzyme design of DdCBEs significantly reduces the risk of off-target deamination events, a critical consideration in clinical applications where unintended mutations could have severe consequences.
Limitations and Challenges
Despite the promising outcomes, several challenges remain before mitochondrial base editing can be translated into clinical practice:
Delivery Efficiency: While AAV vectors are effective in mouse models, scaling up for human applications presents challenges in terms of tissue specificity, immunogenicity, and vector dosage.
Heteroplasmy Dynamics: The distribution and replication of edited versus unedited mtDNA in different tissues and over time must be carefully monitored, as a reversion to pathogenic heteroplasmy levels could undermine therapeutic efficacy.
Long-Term Safety: The long-term effects of base editing in mitochondria, including potential off-target modifications and immunological responses, require extensive investigation.
Regulatory Hurdles: As with any gene therapy, stringent regulatory oversight is necessary to ensure that the risks are minimized and that the therapeutic benefits justify the potential adverse effects.
Broader Implications for Mitochondrial Medicine
The implications of this work extend beyond the correction of a single mutation. The principles underlying mitochondrial base editing can be applied to a broad spectrum of pathogenic mtDNA mutations, offering hope for the treatment of a range of mitochondrial disorders. Additionally, the insights gained from these studies may inform the development of novel therapeutic strategies for age-related and degenerative diseases where mitochondrial dysfunction plays a contributory role.
Future Directions and Clinical Translation
Future research will likely focus on several key areas:
Optimization of Delivery Systems: Advances in vector technology, including the development of novel serotypes or non-viral delivery systems, may enhance the efficiency and specificity of mitochondrial gene delivery in human tissues.
Expansion of Editing Capabilities: Efforts to expand the repertoire of nucleotide conversions ”beyond C-to-T transitions will broaden the therapeutic applicability of mitochondrial base editing.
Combination Therapies: Integrating base editing with other therapeutic modalities, such as mitochondrial biogenesis enhancers or antioxidants, could provide a synergistic approach to treating complex mitochondrial diseases.
Rigorous Preclinical Studies: Extensive long-term studies in larger animal models will be essential to evaluate the durability and safety of mitochondrial base editing before clinical trials can be initiated.
Ethical and Regulatory Considerations
The promise of mitochondrial gene therapy is accompanied by significant ethical and regulatory challenges. The manipulation of mitochondrial genomes raises questions about germline modifications and the potential for unintended consequences in future generations. Therefore, robust ethical frameworks and regulatory policies must be established to govern the application of these technologies. Public and professional discourse will play an essential role in balancing the therapeutic potential of mitochondrial base editing against the ethical imperatives of safety and informed consent.
Conclusion
In summary, the correction of a pathogenic mtDNA mutation in mice via base editing represents a transformative advancement in the field of mitochondrial genetics and gene therapy. By leveraging the precision of DdCBE technology, researchers have demonstrated that it is possible to achieve a significant heteroplasmy shift, restore mitochondrial function, and ameliorate clinical symptoms in a preclinical model. Although challenges remain particularly in the realms of delivery, long-term safety, and ethical oversight the potential of this approach to offer curative treatments for mitochondrial diseases is both compelling and inspiring.
The implications of this work are vast, suggesting that with further refinement and rigorous clinical testing, mitochondrial base editing could soon transition from a promising experimental strategy to a viable therapeutic option for patients suffering from otherwise intractable mitochondrial disorders. It is the hope of the scientific and medical communities that continued research in this area will ultimately lead to a paradigm shift in the treatment of mitochondrial diseases, providing relief to countless individuals and their families worldwide.
References
Note: For the purposes of this essay, references have been synthesized from general knowledge in the field. In an academic context, proper citations to primary literature and review articles should be provided.
1. Wallace, D. C. (2010). Mitochondrial DNA mutations in disease and aging. Environmental and Molecular Mutagenesis, 51(5), 440450.
2. Gammage, P. A., Rorbach, J., Vincent, A. I., Rebar, E. J., & Minczuk, M. (2018). Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Molecular Medicine, 10(5), e8779.
3. Mok, B. Y., de Moraes, M. H., Zeng, J., Bosch, D. E., Kotrys, A. V., Raguram, A., … & Liu, D. R. (2020). A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature, 583(7817), 631637.
4. Payne, B. A. I., & Chinnery, P. F. (2015). Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochimica et Biophysica Acta (BBA) – Bioenergetics, 1847(11), 13871392.
The development of mitochondrial base editing, as exemplified by this study, represents a significant stride toward addressing the fundamental causes of mitochondrial disease. It is an exciting time for mitochondrial research, and it is the opinion of this author that the continued integration of molecular innovation with rigorous clinical investigation will ultimately yield effective and safe treatments for these devastating conditions.